Self-Inhibition of Synthesis and Antigen Presentation by Epstein-Barr Virus-Encoded EBNA1The glycine-alanine repeat domain (GAr) of Epstein-Barr virus–encoded nuclear antigen 1 (EBNA1) prevents major histocompatibility complex (MHC) class I–restricted presentation of EBNA1 epitopes to cytotoxic T cells. This effect has previously been attributed to the ability of GAr to inhibit its own proteasomal degradation. Here we show, both in vitro and in vivo, that GAr also inhibits messenger RNA translation of EBNA1 in cis and that this effect can be distinguished from its effect on proteasomal degradation. Hence, inhibition of messenger RNA translation, but not protein degradation, is essential to prevent antigen presentation on MHC class I molecules. Thus, by minimizing translation of the EBNA1 transcript, cells expressing EBNA1 avoid cytotoxic T cell recognition. At the same time, blocking degradation maintains the EBNA1 expression level.
1 Division of Molecular Physiology, Faculty of Life Sciences, University of Dundee, Dundee DD1 5EH, UK. * To whom correspondence should be addressed. E-mail: [email protected]
Viruses have evolved numerous mechanisms to evade the host immune system (1, 2). A well-documented example is provided by EBNA1, which is expressed in all Epstein-Barr virus (EBV)–carrying cells and is essential for maintaining viral replication (3). In certain types of cells, such as Burkitt's lymphoma, expression of EBV antigens is restricted to EBNA1 only (4, 5). CD8+ cytotoxic T lymphocytes (CTLs) specific for EBNA1-derived peptides are generated via cross-priming and can readily be isolated from EBV-infected individuals (6–8). Although these CTLs can kill human leukocyte antigen–compatible cells loaded with exogenous EBNA1-derived peptides, they fail to recognize cells expressing endogenous full-length EBNA1 (8). This is a result of a failure to present antigens and can be corrected after deletion of the Gly-Ala repeat domain (GAr) of EBNA1 (8). Furthermore, fusion of GAr to an unrelated protein can confer inhibition of presentation to CTLs (9). Previous studies have also shown that GAr increases the half-life of EBNA1 (and other proteins to which it is fused in cis) by inhibiting degradation via the proteasomal pathway. This self-inhibition of degradation has been proposed as the mechanism by which MHC class I presentation of EBNA1-derived peptides is inhibited (10, 11). However, this model leaves questions concerning the mechanisms by which EBNA1-expressing cells avoid CD8+ T cell recognition unanswered. For example, an increase in the half-life conferred by GAr should result in a higher steady-state level of full-length EBNA1 than of EBNA1 with a deleted GAr. In fact, this is not observed and EBNA1 does not accumulate to a high level in EBV-infected B cells, nor in cells expressing exogenous EBNA1 (8, 10). Furthermore, by depending solely on inhibition of proteasomal degradation of full-length EBNA1 as a means of avoiding CTL recognition, GAr would not be capable of preventing presentation of peptides from the EBNA1 transcript that are derived through rapid degradation of so-called defective ribosomal products (DRiPs) (12, 13). Such DRiPs have been estimated to make up at least 30% of newly synthesized mRNA translation products, and are thus considered a potentially important source of peptides for presentation by MHC class I molecules (14, 15). These questions prompted us to further investigate the role of the GAr sequence in preventing EBNA1 from producing antigenic peptides.
Contrary to expectations, the EBNA1 protein steady-state level was increased in cells that expressed an EBNA1 with a deleted GAr (EB
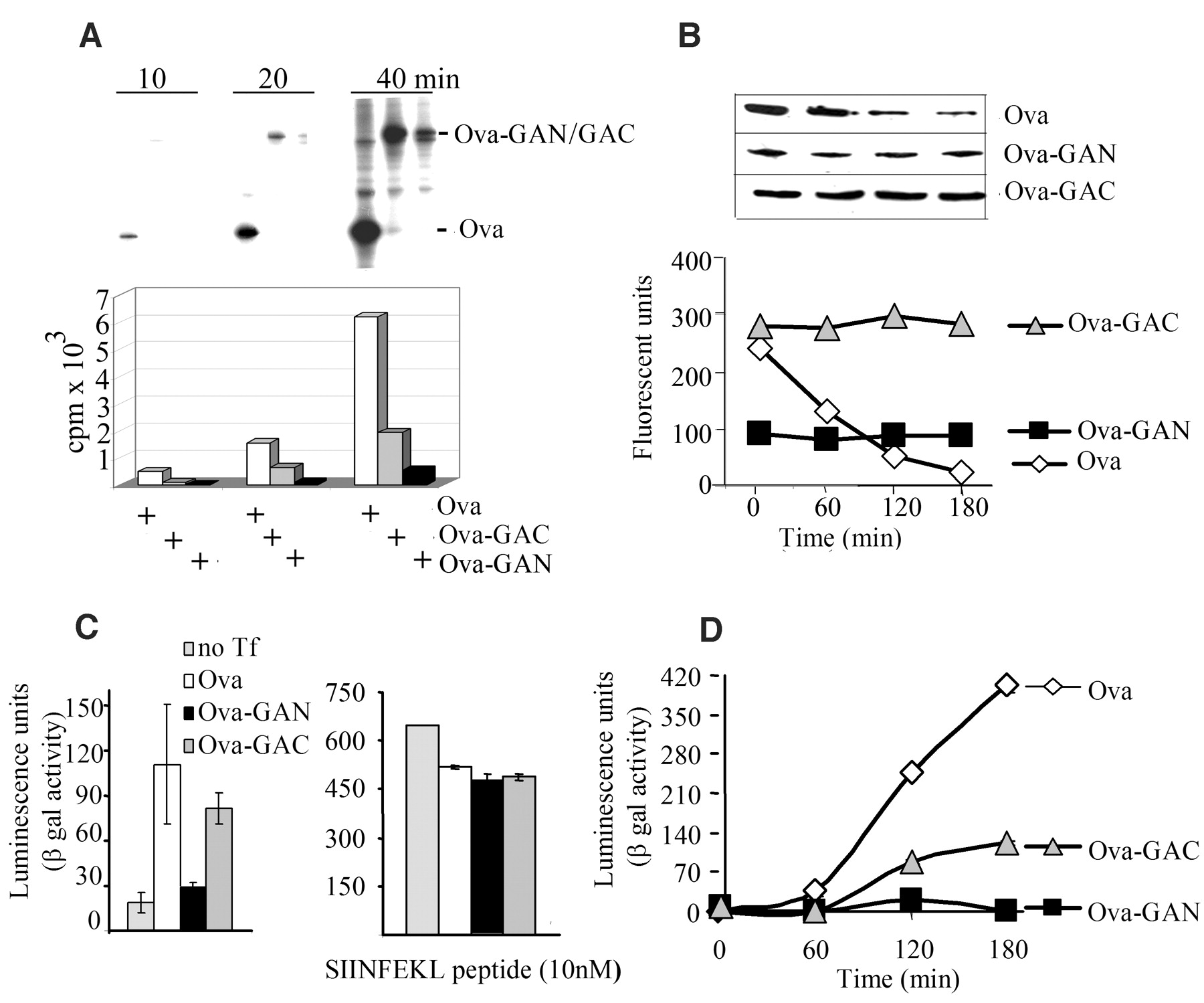
The double-stranded RNA–activated kinase (PKR) phosphorylates the translation initiation factor eIF2 To determine whether other parts of the EBNA1 transcript might contribute to the inhibition of translation, which could help to explain the location-dependent effect of GAr, we fused GAr to the C terminus or to the N terminus of an ovalbumin (Ova) sequence that lacks the N-terminal 50–amino acid signal sequence (Ova-GAC and Ova-GAN, respectively) (Fig. 2E). These Ova transcripts were translated at levels similar to the corresponding EBNA1 transcripts; hence, no other parts of the EBNA1 mRNA are required for GAr to block translation. We also fused a shortened GAr sequence (corresponding to 20 amino acids) to the C terminus or to the N terminus of the Ova transcript (Ova-Cpep and Ova-Npep, respectively) (Fig. 2E). This short sequence had a reduced effect on translation relative to full-length GAr when located at the N terminus and displayed no effect on translation when fused to the C terminus of the Ova mRNA.
The observation that the effect of GAr is dependent on its position within the coding region led us to consider the possibility that the GAr peptide sequence was capable of inhibiting its own synthesis. We reasoned that if this assumption were correct, the GAr peptide might inhibit mRNA translation by interfering with the activity of a ribosomal factor, and translation of the GAr transcript might be stimulated if the GAr peptide were hindered from such an interaction. To test this idea, we purified GAr sequence-specific antibodies derived from EBV-positive human sera and from rabbits immunized with GAr peptides (17), confirming GAr specificity by immunoblotting (Fig. 2F, upper panel) and enzyme-linked immunosorbent assay (18). When these antibodies were added to IVT assays, they increased the translation of the EBNA1wt transcript but had no effect on translation of the EB We next looked at the effect of GAr on intracellular protein synthesis. Cells expressing Ova or Ova-derived fusion proteins carrying the GAr sequence in the C terminus or N terminus were pulse-labeled with [35S]methionine for 10, 20, and 40 min. Figure 3A shows that, as in vitro, GAr-mediated inhibition of synthesis in vivo depended on the location of the GAr sequence within the protein.
Taken together, these results may explain the paradox of why removal of the GAr sequence from EBNA1 has only a limited effect on EBNA1 expression in cells. Apparently, the increase in synthesis of EBNA1 after removal of GAr is almost completely compensated for by the subsequent increase in the rate of degradation. The idea that GAr-mediated inhibition of proteasomal degradation of full-length EBNA1 is sufficient to avoid EBNA1-derived peptides from being processed and loaded onto MHC class I molecules (9, 11) is not in accordance with the DRiP hypothesis (13, 14, 20). We therefore tested directly whether the inhibition of proteasomal degradation was sufficient to protect GAr chimeric proteins from being processed and presented on MHC class I molecules. To do this, we took advantage of the fact that GAr-mediated self-inhibition of mRNA translation is dependent on the location of GAr within the protein, whereas inhibition of proteasomal degradation is not (Fig. 3, A and B) (11). Thus, by changing the location of the GAr sequence, the rate of translation could be altered while maintaining the rate of degradation of the full-length protein. We used the Ova-GAr fusion constructs to compare the effects of inhibition of protein degradation (Ova-GAC) with those of the combined inhibition of degradation and synthesis (Ova-GAN) on generating antigenic peptides. Kb-positive EL4 cells expressing Ova-GAN, Ova-GAC, or Ova were incubated with the B3Z CD8+ T cell clone specific for the Ovaderived SIINFEKL peptide (Ser-Ile-Ile-Asn-Phe-Glu-Lys-Leu, residues 257 to 264) for 20 hours (21). Expression of Ova and Ova-GAC led to T cell stimulation, whereas expression of Ova-GAN did not (Fig. 3C, left panel). To ensure that GAr did not affect the MHC class I antigen presentation pathway, we exposed cells that expressed the different Ova transcripts to exogenous SIINFEKL peptide before incubating them with the B3Z cells. Under these conditions, there was no difference in the capacity of GAr-expressing EL4 cells to present SIINFEKL peptides (Fig. 3C, right panel). We also compared the rate of production of SIINFEKL peptide from the different Ova constructs by treating Ova-expressing EL4 cells with brefeldin A (BFA) for 12 hours, followed by the addition of the reversible proteasome inhibitor MG132. An increase in the presentation of SIINFEKL peptide was observed over a 3-hour time period after BFA and MG132 were removed from cells expressing Ova or Ova-GAC but not Ova-GAN (Fig. 3D). A similar increase in SIINFEKL presentation was observed in cells given fresh medium after being treated only with MG132 for 4 hours (18). The rate of SIINFEKL production from the Ova-GAC construct is about one-third that of Ova, which correlates with the difference in the rate of mRNA translation of these two transcripts. Hence, we conclude that inhibition of proteasomal degradation by GAr is not sufficient to prevent presentation of epitopes from GAr chimeras to the MHC class I pathway, and that the increase in antigenic peptide production observed from the Ova-GAC transcript (relative to that of Ova-GAN) reflects its higher rate of mRNA translation. There are many examples of how viruses evade immune recognition of host cells, but EBV appears unique in that its latent form of existence in cell types that express EBNA1 only (such as Burkitt's lymphoma cells) is reliant on the GAr peptide motif to protect the host cell from immune recognition. Even though it is clear that the ability to block its own degradation is an important part of the viral strategy to control the expression level of EBNA1, these results show that protection of the full-length protein from proteasomal processing is not sufficient to block antigen presentation. Instead, our results show how EBV has evolved a mechanism to avoid immune surveillance of EBNA1-expressing latent host cells. This is a beautiful example of an adaptation to the rules of the DRiP model, in that the most effective way to prevent the production of potentially antigenic products from the EBNA1 mRNA is to suppress its translation. A slower rate of translation might ensure that peptides that are translated out of frame (or that carry nonfunctional GAr sequences for other reasons) will not be produced from the EBNA1 transcript. Such a reassurance could not be achieved by simply lowering the rate of transcription of the gene encoding EBNA1. And because EBNA1 has no other known function for interfering with antigen presentation, inhibition of mRNA translation is clearly an effective strategy. The identification of a virus-encoded protein such as EBNA1 that minimizes MHC class I antigen presentation by inhibiting its own synthesis adds further support to the importance of rapidly degraded translation products as the main source for antigenic peptides. This example of mRNA translation regulation by a peptide sequence within the encoded polypeptide is unusual, and there are to our knowledge few, if any, similar examples (22–24). This unusual self-regulating mechanism of protein synthesis most likely reflects the fact that GAr carries out a unique dual function, as an inhibitor of both ribosomal and proteasomal activity. Thus, to minimize translation while maintaining a functional expression level of EBNA1, GAr appears to adapt to and explore as yet unidentified common mechanisms that control both proteasomal and ribosomal activity.
Supporting Online Material www.sciencemag.org/cgi/content/full/301/5638/1371/DC1 Materials and Methods
8 July 2003; accepted 6 August 2003
Related articles in Science:
Volume 301, Number 5638, Issue of 5 Sep 2003, pp. 1371-1374. Copyright © 2003 by The American Association for the Advancement of Science. All rights reserved. |
 GA) relative to cells expressing the wild type (EBNA1wt) (
GA) relative to cells expressing the wild type (EBNA1wt) (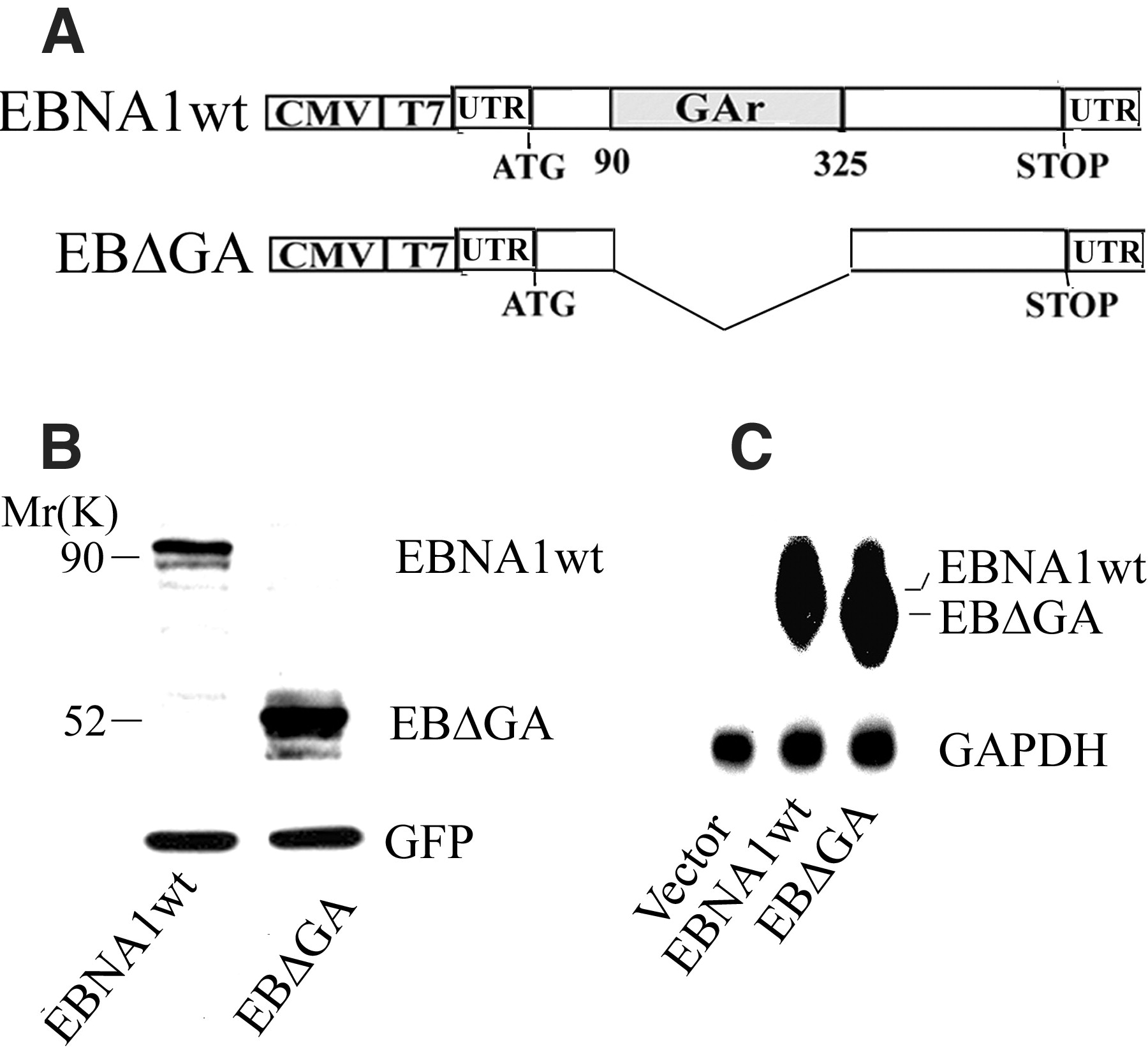
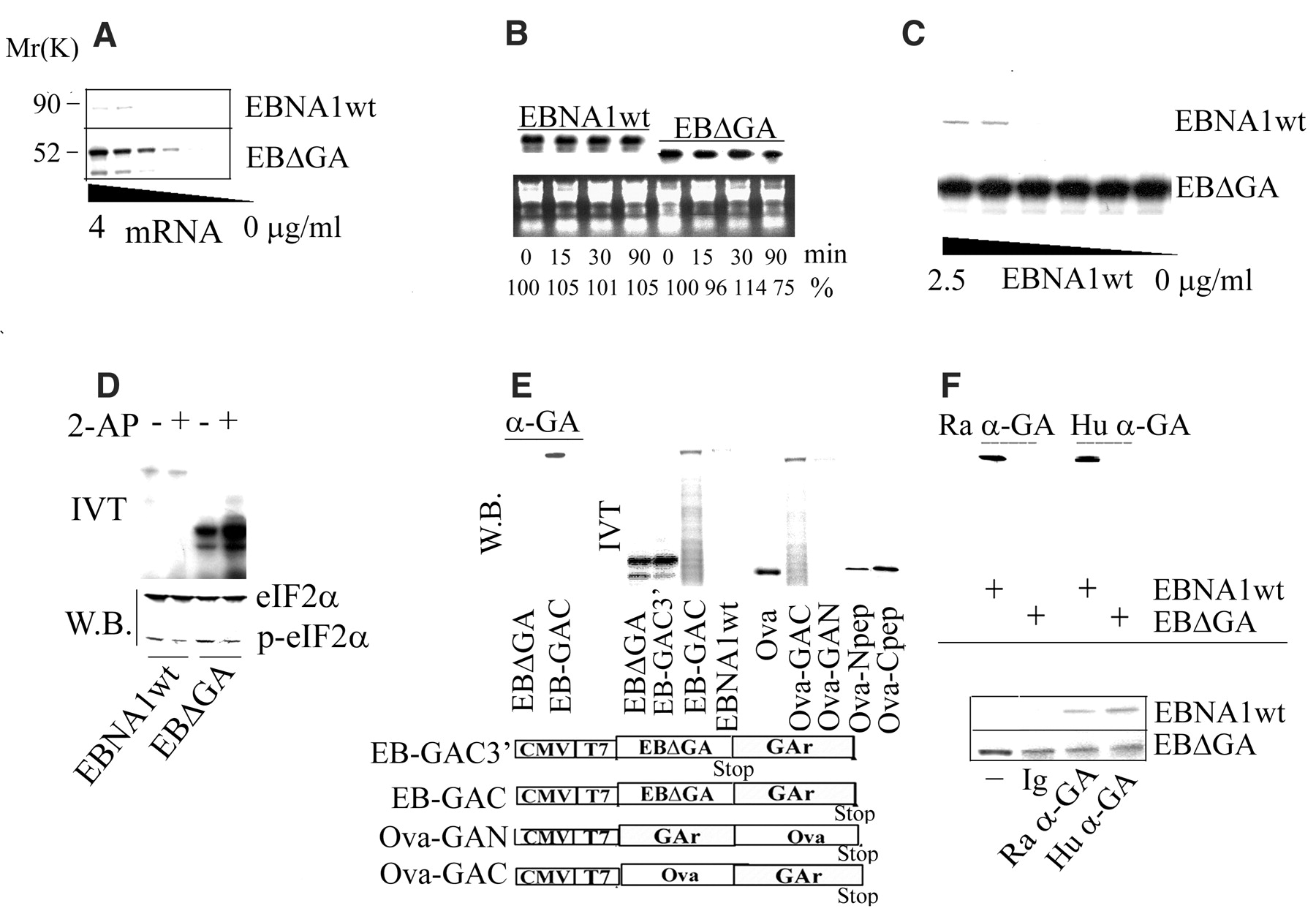
 11 increase in EBNA1 translation at 4 µg/ml mRNA. (B) EBNA1wt and EB
11 increase in EBNA1 translation at 4 µg/ml mRNA. (B) EBNA1wt and EB [32P]uridine triphosphate in vitro and added to rabbit reticulocyte lysate for the indicated lengths of time. Total RNA was isolated and visualized by ultraviolet light (lower panel), and the stability of EBNA1 mRNAs was determined by phosphoimager measurement of EBNA1wt and EB
[32P]uridine triphosphate in vitro and added to rabbit reticulocyte lysate for the indicated lengths of time. Total RNA was isolated and visualized by ultraviolet light (lower panel), and the stability of EBNA1 mRNAs was determined by phosphoimager measurement of EBNA1wt and EB